Tool usage (CLI)
There are two main commands in CNSistent: cns and segment. The segment command is used to create segmentations, while the cns command is used to process CNS data, based on segments.
The command line interface uses the following pattern:
cns [command] cns_file_path [options]
The following commands are available (see details below):
align: Adds missing segments to the CNS data to match the assembly (fills gaps with NaNs).
infer: Infers values for NaNs in the CNS data.
impute: Imputes missing values in the CNS data - combination of align and infer.
coverage: Calculates coverage for aligned (but not imputed) CNS data.
ploidy: Calculates aneuploidy for CNS data (NaNs are ignored).
breakage: Creates a clustering of breakpoints.
aggregate: Creates bins for CNS data.
The cns_file_path must point to a CNS file as described in the following section.
The segment command uses the following pattern:
segment [options]
The output of the segment command is a BED file with the segments, which can be used as input to the cns command using the --segments argument.
Data format
The tool expects an unprocessed copy number dataset in the form of a TSV file with the following column scheme: sample_id, chrom, start, end, [*_cn].
The
sample_idis the identifier of the sample,chromis the name of the chromosome,startandendare the start and end positions of the segment,[*_cn]is typically one or two copy number segments.
E.g.:
sample_id |
chrom |
start |
end |
CN1 |
CN2 |
s1 |
chr19 |
1000000 |
3000000 |
1 |
|
s1 |
chr19 |
3000000 |
12000000 |
1 |
1 |
s1 |
chr19 |
12000000 |
14000000 |
1 |
|
s1 |
chr19 |
14000000 |
21000000 |
3 |
1 |
s1 |
chr19 |
21000000 |
25000000 |
3 |
|
s1 |
chr19 |
28000000 |
58500000 |
3 |
|
s2 |
chr19 |
1000000 |
24000000 |
2 |
|
s2 |
chr19 |
29000000 |
58000000 |
0 |
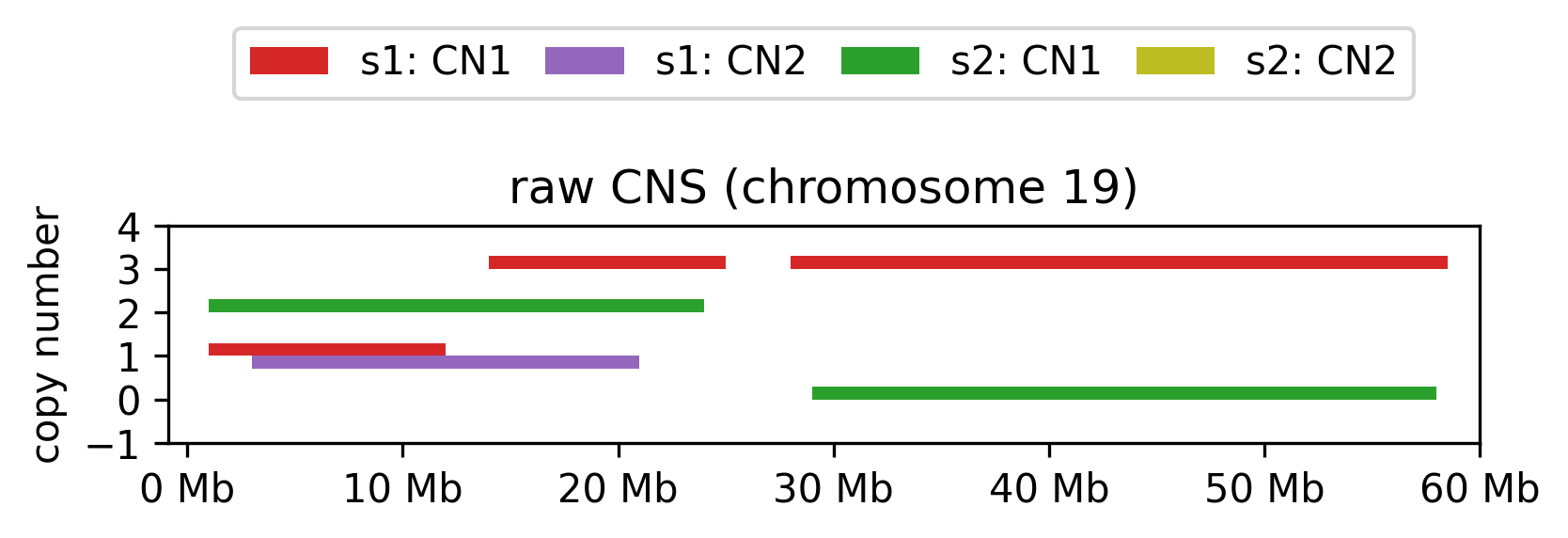
Raw copy number data for each sample and allele.
Note
To conform with the standard practice, the start and end positions are 1-based, and the end position is inclusive. However, for the sake of sanity of the author, internally these are converted to 0-based, and the end position is exclusive.
Input format
The canonical format of the input data is sample_id, chrom, start, end for the segment positions and
major_cn, minor_cn for the copy number values if there are two value ordered alleles, hap1_cn, hap2_cn if there are two unordered alleles, and total_cn if there is only one value for the copy number.
The following alternate names are also parsed`, CASE INSENSITIVE:
sample_id:sample, id, sampleid, sample-id, sample_name, samplename, sample-namechrom:chromosome, chrstart:being, startpos, start_pos, start-pos, chromstart, chrom_start, chrom-startend:stop, endpos, end_pos, end-pos, chromend, chrom_end, chrom-endCN column: Has
cn | hap | major | minor | total | allelein the name.
Samples file
To know which sex chromosomes are expected in each sample, it is possible to provide a samples file using the --samples argument with the following format:
sample_id |
sex |
s1 |
xy |
s2 |
xx |
If this is not provided, the sex is determined by presence of the Y chromosome in the data.
If the samples files is provided, only the samples listed in the file are processed, even if the CNS file would have more samples.
Some of the commands create samples information, for example ploidy. It is possible to use the same file as both input and output.
Segmentation files
A calculation can be restricted to certain segments by providing a BED file with the segments to be used. The BED file must have the following columns: chrom, start, end.
chrom |
start |
end |
chr1 |
1000000 |
2000000 |
chr1 |
3000000 |
4000000 |
… |
The segment command creates a segmentation file.
Single sample input
If you aim to process just a single sample, you can format it for input using the following command:
awk 'BEGIN{FS="[ \t]+";OFS="\t"} {print "sample1", $1, $2, $3}' yourfile.txt | sed 'sample_id\tchrom\tstart\tend' > modified_file.tsv
Commands
Common arugments
--samples: path to the samples (TSV) file.--segments: path to the segments (BED) file.--out: path to the output file. Default iscns.out.tsv.--assembly: assembly version to use. Default ishg19.--cncols: If the CN columns do not conform to the naming as above, or if there are more columns, one or two columns can be specified, comma separated, no whitespace e.g.cn1,cn2.--threads: number of threads to use. Default is 1.--subsplit: will split the data in multiple blocks and process them in sequence, in case of low memory. Default is 1.--verbose: print progress information.--timeit: times the calculation and writes to
align
Aligns all the segments so that each samples spans the whole reference. The following steps are performed:
Added NaN segments to the telomeres.
Fill gaps in the data with NaN values.
Add missing chromosomes, if they are missing compared to the reference.
Merge neighbouring segments with the same copy numbers (or NaNs). Both minor and major must match.
infer
Replaces any NaNs in the CNS file with the values of the closest neighbouring region that is not NaN. The following steps are performed:
Assign telomeres the values of the closest neighbouring region is not NaN.
Split the gaps and to each side, assign the values of the closes neighbouring region that is not NaN, in the direction from the center towards the side (see example below).
If a whole chromosome is missing, or declared as NaN, its assigned to 0 for its whole length.
Merge neighbouring segments with the same copy numbers (or NaNs). Both minor and major CN values must match to be merged.
Additional arguments:
--method:extend, diploid, zero. The method to use for imputation. Default isextend.extend: extends the closest non-NaN value to the telomeres and gaps.diploid: assigns the expected diploid value (1 for each allele on autosomes and female X chromosomes, 1/0 for male X and Y chromosomes).zero: assigns 0 to all NaN values.
impute
Combines the align and infer commands to create an CNS file.
Additional arguments:
--method:extend, diploid, zero. Same as for theinfercommand. Default isextend.
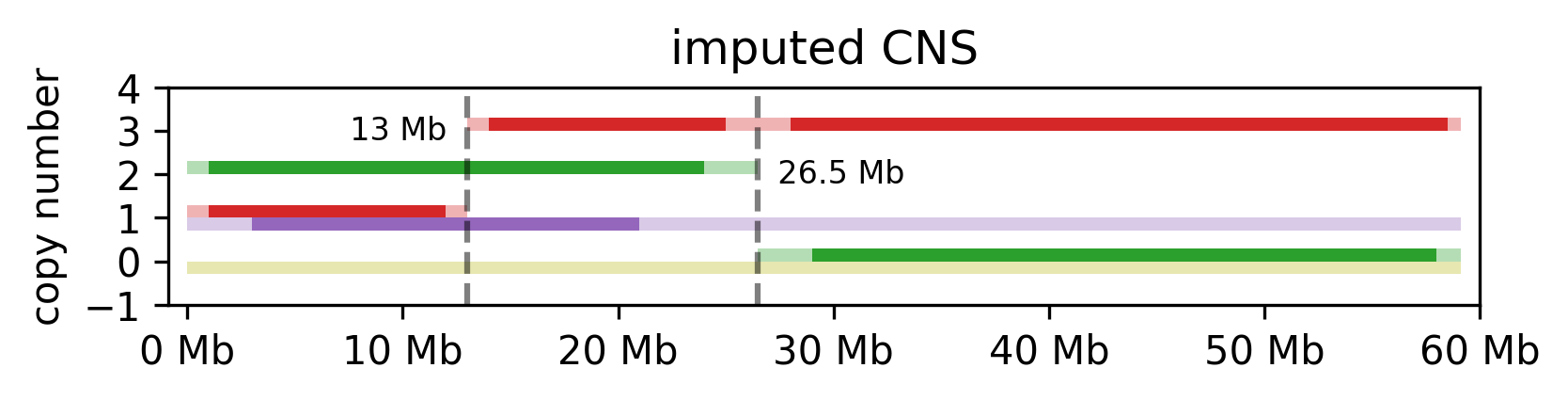
coverage
Calculates the coverage of the CNS file. The coverage is calculated as the fraction of the genome that has a CN value assigned.
Note
Coverage should be run on a aligned, but not inferred dataset.
Note
For all sample statistics, the values are calculated for autosomes, sex chromosomes, and the total genome, with the values being suffixed with _aut, _sex, _tot, respectively. If sex chromosomes are missing from data altogether, only _aut values are calculated.
The following statistics are calculated and stored in a samples file:
sex:xyfor male,xxfor female. If this information is not specified,xyis used if and only ifchrYis present in the sample.chrom_count: the number of autosomes that had any CN values assignedchrom_missing: the list of chromosomes that have no CN values assignedcoverage_{any,all}_{aut,sex,all}: proportion of the genome that has a CN value assigned,anyfor either allele (one allele is sufficient),allfor both alleles (both alleles are required),
feature |
s1 |
s2 |
coverage (any) |
0.966139 |
0.928091 |
coverage (all) |
0.285566 |
0.0 |
ploidy
Calculates the portions of the genome that are aneuploid, or for absent in case of male sex chromosomes.
Note
ploidy should be run on an imputed dataset.
The following statistics are calculated and stored in a samples file:
loh_{any,hot}_{aut,sex,all}: proportion of the chromosome set that has CN=0 for an allele (any) or both alleles (all).ane_{any,hot}_{aut,sex,all}: proportion of the chromosome set that has CN different from 1 for an allele (any) or both alleles (all). In one column formatanycan’t be established.imb_{CN1,CN2}_{aut,sex,all}: proportion of the chromosome set where one allele has strictly higher CN. In one column format this is not calculated.
feature |
s1 |
s2 |
aneuploidy (all) |
0.0 |
1.0 |
aneuploidy (any) |
0.767977 |
1.0 |
LOH (all) |
0.0 |
0.560374 |
LOH (any) |
0.0 |
1.0 |
imbalance (CN1) |
0.767977 |
0.439626 |
imbalance (CN2) |
0.0 |
0.0 |
breakage
Calculates the number of breaks and the step size between the breaks for the samples.
The following statistics are calculated:
breaks_{CN1,CN2,total_cn}_{aut,sex,all}: the number of breaks in the CN values for the allele.step_{CN1,CN2,total_cn}_{aut,sex,all}: the average step size between the breaks in the CN values for the allele.
feature |
s1 |
s2 |
breaks (CN1) |
1 |
1 |
step (CN1) |
2 |
2 |
breaks (CN2) |
0 |
0 |
step (CN2) |
0 |
0 |
breaks (total) |
1 |
1 |
step (total) |
2 |
2 |
aggregate
Aggregates CN values across segments, creating a consistent segmentation for each sample based on a provided BED file. The --segments argument must be provided for aggregation.
Note
BED file is 0 indexed!
Additional arguments:
--how:mean, min, max, none. The method to aggregate the CN values. Ifnoneis selected, the CN values are not aggregated, but existing segments are masked by the provided segments.
cns aggregate cns.tsv --segments segments.bed
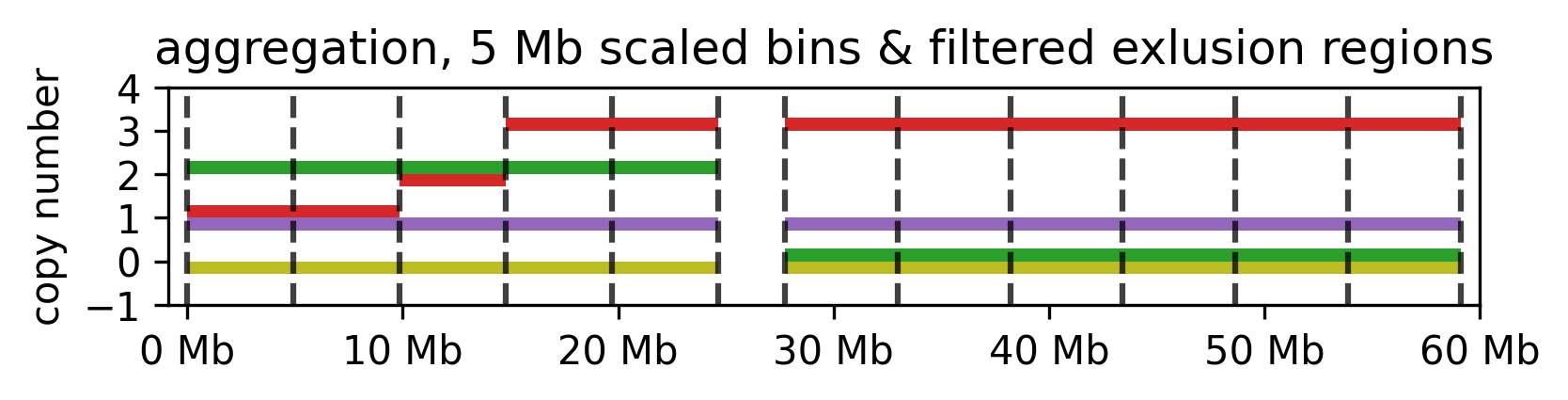
Aggregated CN values for the example segments.
segment
Creates a segmentation scheme.
Note
A CNS file is always expected as input, but if breakpoint merging is not done, this argument is not further used.
Binning can be done on the whole genome, or on selected segments. Additionally, segments can be removed from the dataset before binning. The following steps are performed:
- If
--selectis provided, only the selected segments are used for binning. The segments are selected based on thechrom,start, andendcolumns. The segments can be selected by chromosome, chromosome arm, or chromosome band. 1.1 Values
arms, andbandsare used to select chromosome arms, or chromosome bands, respectively.1.2 If
--filteris provided, segments that are strictly smaller than the value are removed.
- If
- If
--removeis provided, these segments are subtracted from the selection. The segments are removed based on thechrom,start, andendcolumns. The segments can be removed by chromosome, chromosome arm, or chromosome band. 2.1 Value
gapscan be used to remove genomic gaps (regions of low mappability) from the selection. 2.2 If--padis provided, the removal segments are padded on both sides by the given size in base pairs before subtraction. 2.3 If--filteris provided, segments that are strictly smaller than the value are removed both before and after the subtraction process, i.e. a if a remove segment is smaller than the filter value, it is not used in subtraction. If the subtraction results in a segments smaller than the filter, it is likewise not used for binning.
- If
If
--mergeis provided existing breakpoints are merged to match the specified merge distance.If
--splitis provided, the data is binned into segments of the given size. The segments are created by aggregating the CN values of the selected segments.
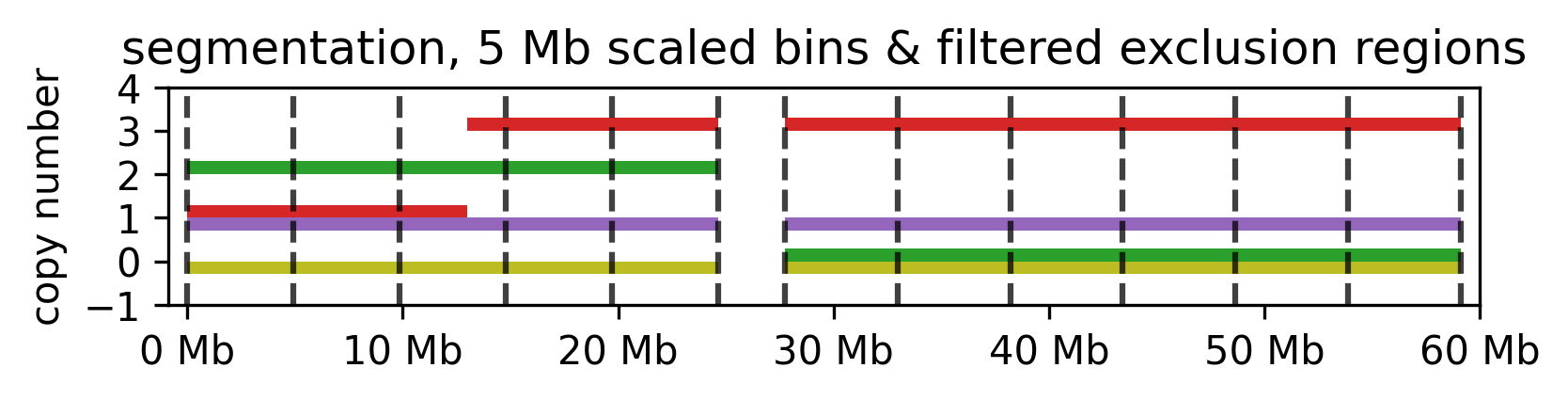
5 mb segmentation of the imputed example segments with gaps removed.