Examples: Plotting
Below are examples of data aggregation and plotting functions that can be used to visualize the data.
[1]:
import cns
import cns.data_utils as cdu
import matplotlib.pyplot as plt
Labeling functions
These functions create background/s labels independent of the data.
[2]:
fig, axs = plt.subplots(5,1, figsize=(15, 15), sharex=True, dpi=50)
cns.plot_chr_bg(axs[0])
cns.plot_cytobands(axs[1], alpha=.25)
cns.add_cytoband_legend(axs[1])
cns.plot_gaps(axs[2], alpha=.25)
cns.add_gap_legend(axs[2])
cns.plot_x_ticks(axs[3])
cns.plot_x_lines(axs[3])
cns.no_y_ticks(axs[3])
Prepare Data
Loads and aggregates the data.
[3]:
pcawg_cns_df = cns.add_total_cn(cdu.load_cns_file("PCAWG_cns_imp.tsv"))
pcawg_cns_df.head()
[3]:
| sample_id | chrom | start | end | major_cn | minor_cn | total_cn | |
|---|---|---|---|---|---|---|---|
| 0 | SP101724 | chr1 | 0 | 27256755 | 2.0 | 2.0 | 4.0 |
| 1 | SP101724 | chr1 | 27256755 | 28028200 | 3.0 | 2.0 | 5.0 |
| 2 | SP101724 | chr1 | 28028200 | 32976095 | 2.0 | 2.0 | 4.0 |
| 3 | SP101724 | chr1 | 32976095 | 33354394 | 5.0 | 2.0 | 7.0 |
| 4 | SP101724 | chr1 | 33354394 | 33554783 | 3.0 | 2.0 | 5.0 |
[4]:
genes_segs = cdu.load_segs_out("segs_COSMIC.bed")
pcawg_1_bin_df = cns.aggregate_by_break_type(cns.cns_head(pcawg_cns_df, 1), 100_000)
pcawg_1_groups_df = cns.group_samples(pcawg_1_bin_df, group_name="100 kb")
pcawg_10_bin_df = cns.aggregate_by_break_type(cns.cns_head(pcawg_cns_df, 10), 1_000_000)
pcawg_10_groups_df = cns.group_samples(pcawg_10_bin_df, group_name="1 mb")
pcawg_50_bin_df = cns.aggregate_by_break_type(cns.cns_head(pcawg_cns_df, 50), 10_000_000)
pcawg_50_groups_df = cns.group_samples(pcawg_50_bin_df, group_name="10 mb")
pcawg_arms_bin_df = cns.aggregate_by_break_type(cns.cns_head(pcawg_cns_df, 10), "arms")
pcawg_arms_groups_df = cns.group_samples(pcawg_arms_bin_df, group_name="arms")
gene_bin_df = cns.aggregate_by_segments(cns.cns_head(pcawg_cns_df, 10), genes_segs)
gene_groups_df = cns.group_samples(gene_bin_df, group_name="COSMIC")
Aggregated into 30363 CNS.
Aggregated into 30399 CNS.
Aggregated into 15256 CNS.
Aggregated into 462 CNS.
Aggregated into 7220 CNS.
Plots
[5]:
fig, ax = plt.subplots(1, 1, figsize=(10, 2), dpi=75)
cns.plot_lines(ax, pcawg_50_groups_df, "major_cn", "blue", "MAJOR PCAWG BLUE", .5, 2)
ax.legend()
[5]:
<matplotlib.legend.Legend at 0x1c470c49d50>
[6]:
fig, ax = plt.subplots(1, 1, figsize=(10, 2), dpi=75)
cns.plot_steps(ax, pcawg_50_groups_df, "major_cn", "blue", "MAJOR PCAWG BLUE", .5, 2)
ax.legend()
[6]:
<matplotlib.legend.Legend at 0x1c46c92d3d0>
[7]:
fig, ax = plt.subplots(1, 1, figsize=(10, 2), dpi=75)
cns.plot_dots(ax, gene_groups_df, "major_cn", "blue", "MAJOR PCAWG BLUE", .5, 1)
ax.legend()
[7]:
<matplotlib.legend.Legend at 0x1c470558b50>
[8]:
fig, ax = plt.subplots(1, 1, figsize=(10, 2), dpi=75)
cns.plot_bars(ax, pcawg_arms_groups_df, "major_cn", "blue", "MAJOR PCAWG BLUE", .5)
ax.legend()
[8]:
<matplotlib.legend.Legend at 0x1c47f812690>
[9]:
fig, ax = plt.subplots(1, 1, figsize=(10, 2), dpi=75)
cns.plot_steps(ax, pcawg_arms_groups_df, "major_cn", "blue", "MAJOR PCAWG BLUE", .5)
ax.legend()
[9]:
<matplotlib.legend.Legend at 0x1c470558e50>
[10]:
fig, ax = plt.subplots(figsize=(10, 2), dpi=75)
cns.plot_heatmap(ax, pcawg_arms_bin_df, "major_cn", max_cn = 16)
[10]:
<Axes: >
Figures
[11]:
cns.fig_lines(pcawg_50_groups_df, cn_columns="total_cn", colors="blue");
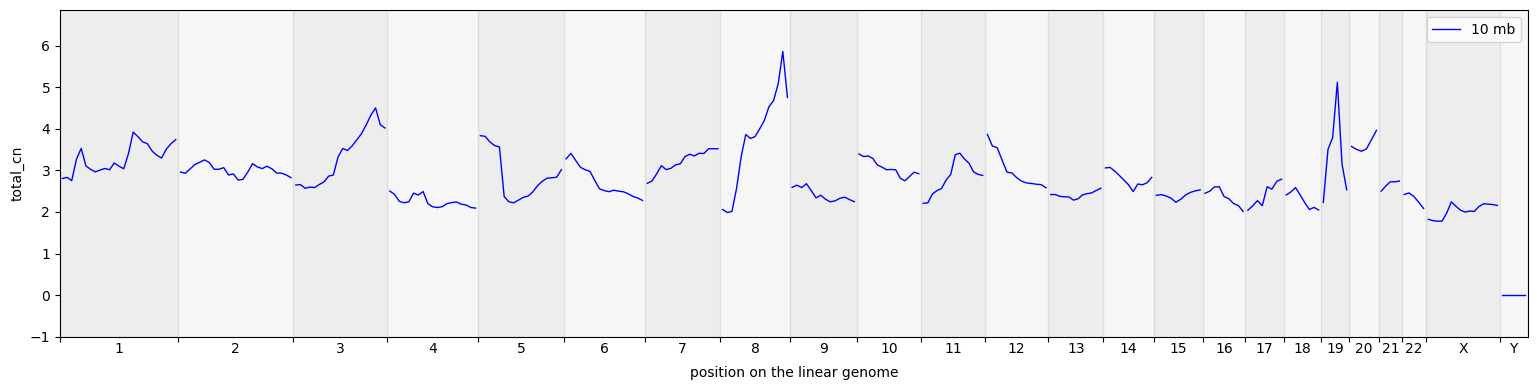
[12]:
cns.fig_steps(pcawg_50_groups_df, cn_columns="total_cn");
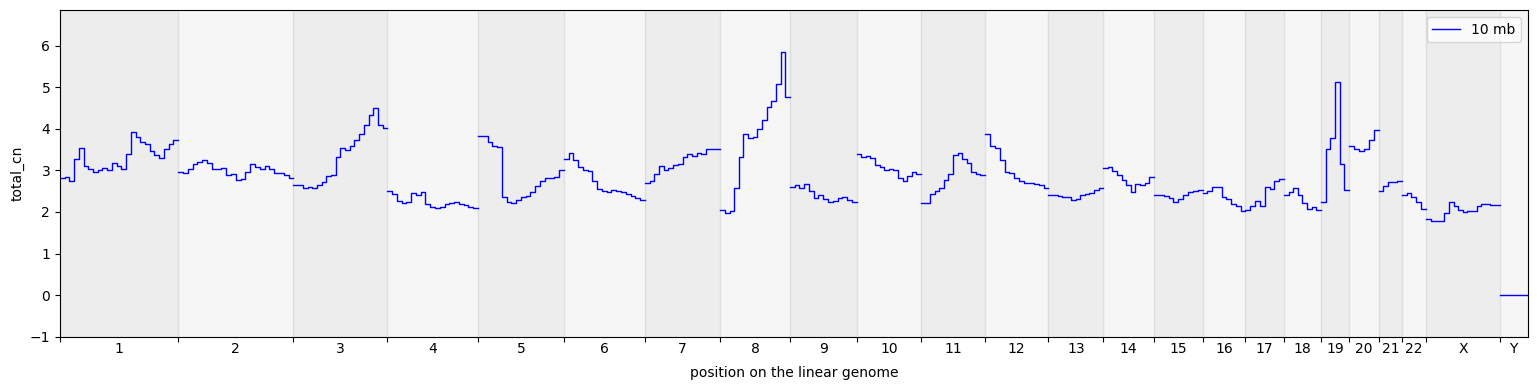
[13]:
groups_df = cns.stack_groups([pcawg_10_groups_df, pcawg_50_groups_df, pcawg_arms_groups_df])
cns.fig_lines(groups_df, cn_columns=["minor_cn", "major_cn"]);
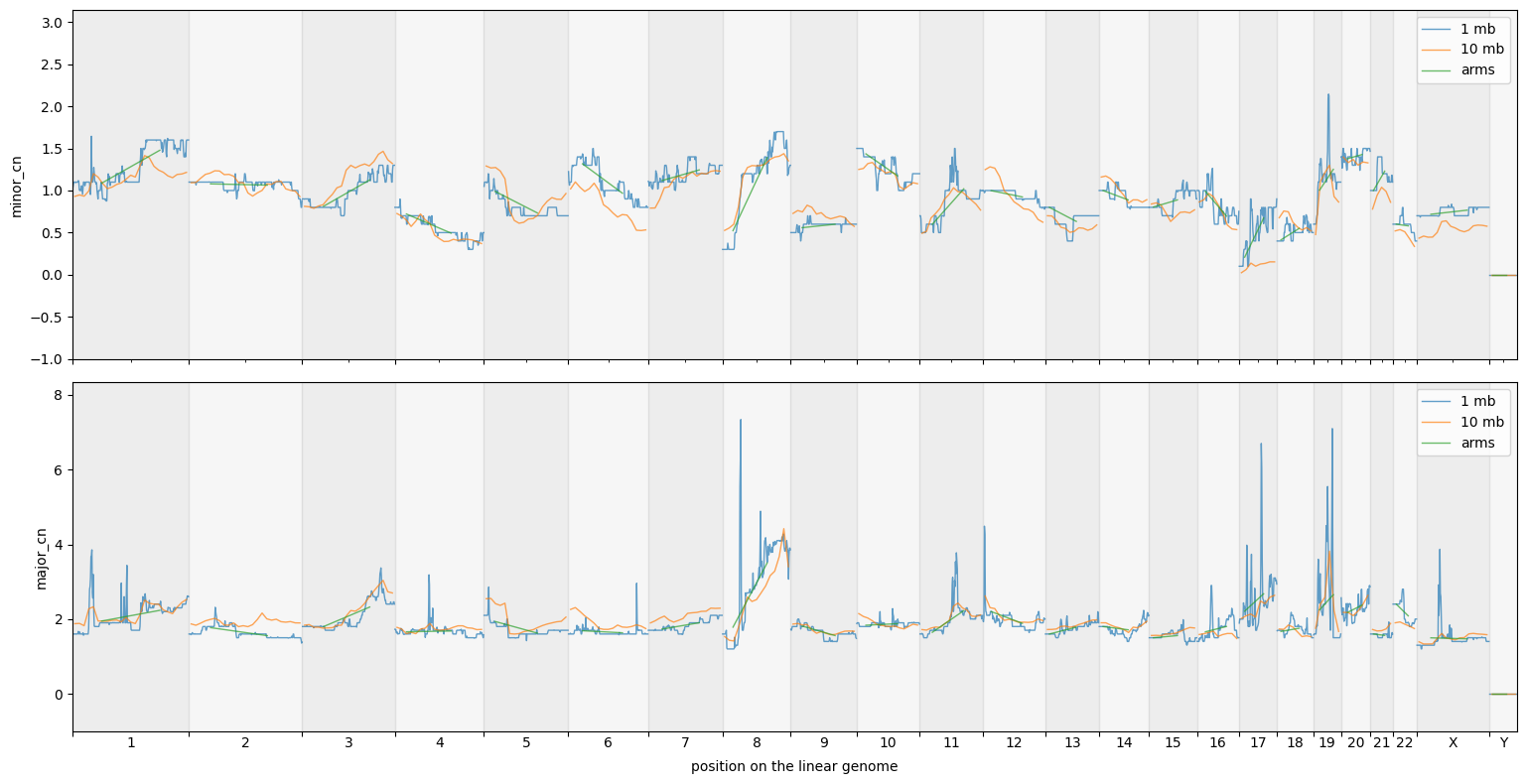
[14]:
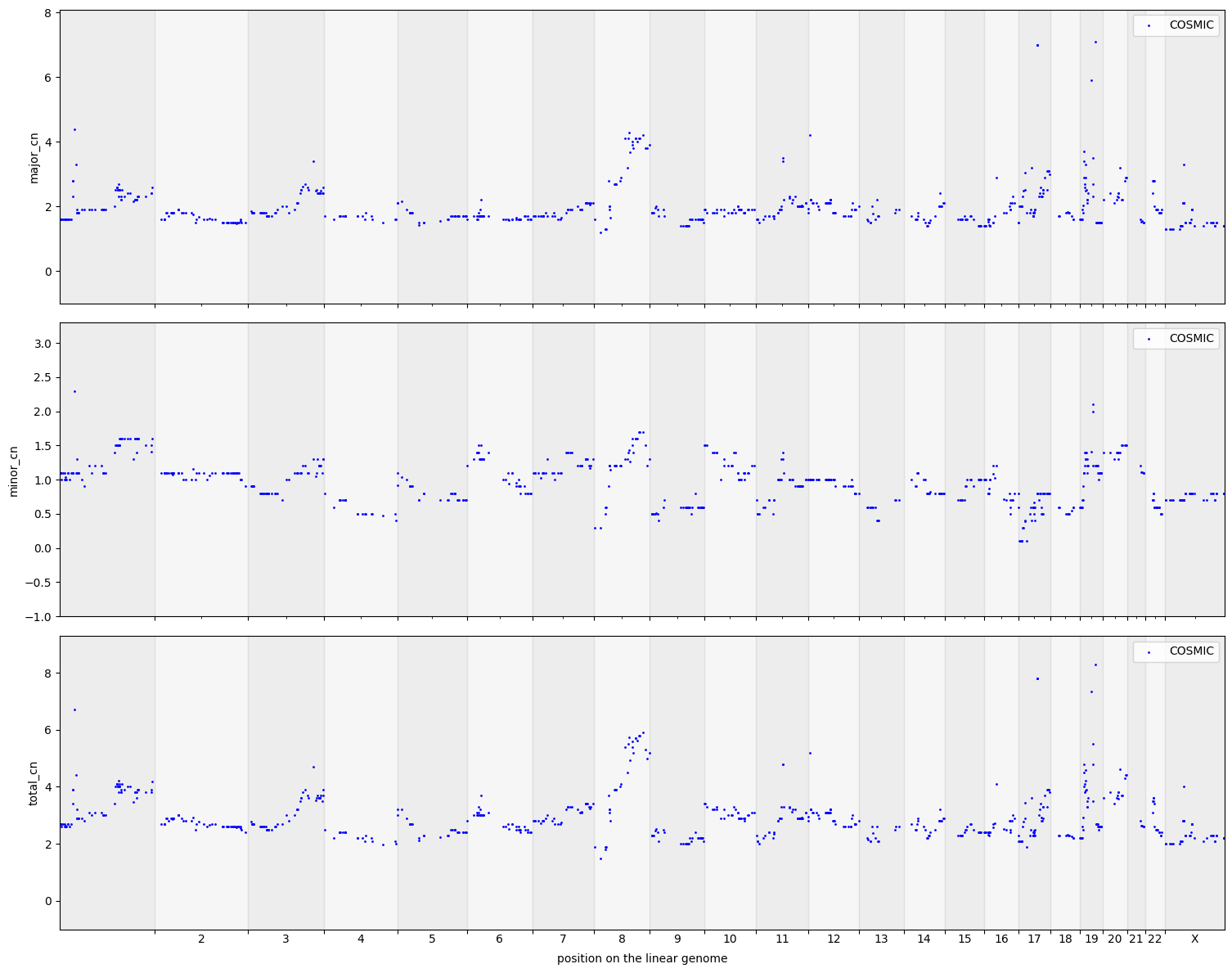
cns.fig_dots(gene_groups_df, cn_columns="total_cn");
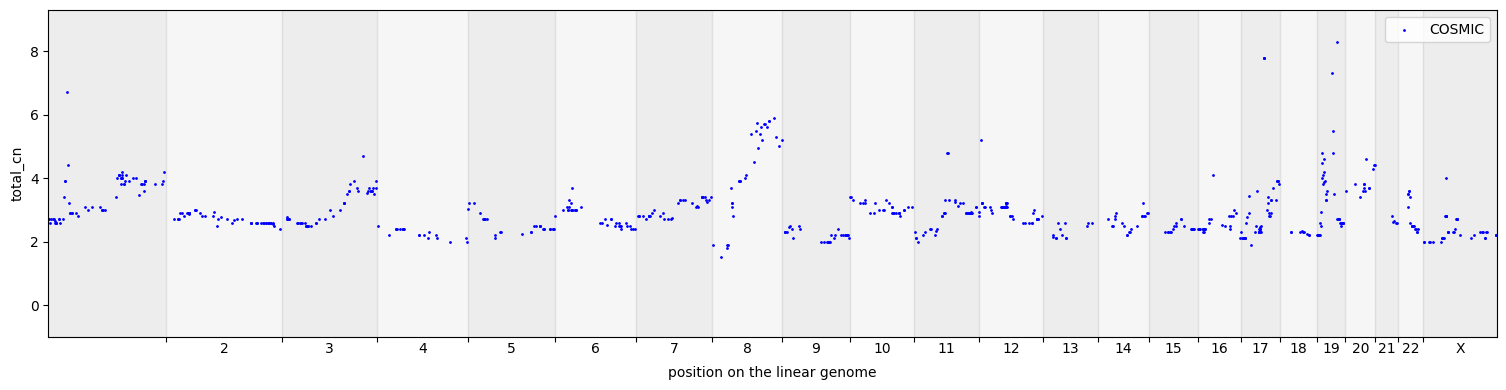
[15]:
cns.fig_dots(gene_groups_df)
[15]:
(<Figure size 1516.62x1200 with 3 Axes>,
array([<Axes: ylabel='major_cn'>, <Axes: ylabel='minor_cn'>,
<Axes: xlabel='position on the linear genome', ylabel='total_cn'>],
dtype=object))
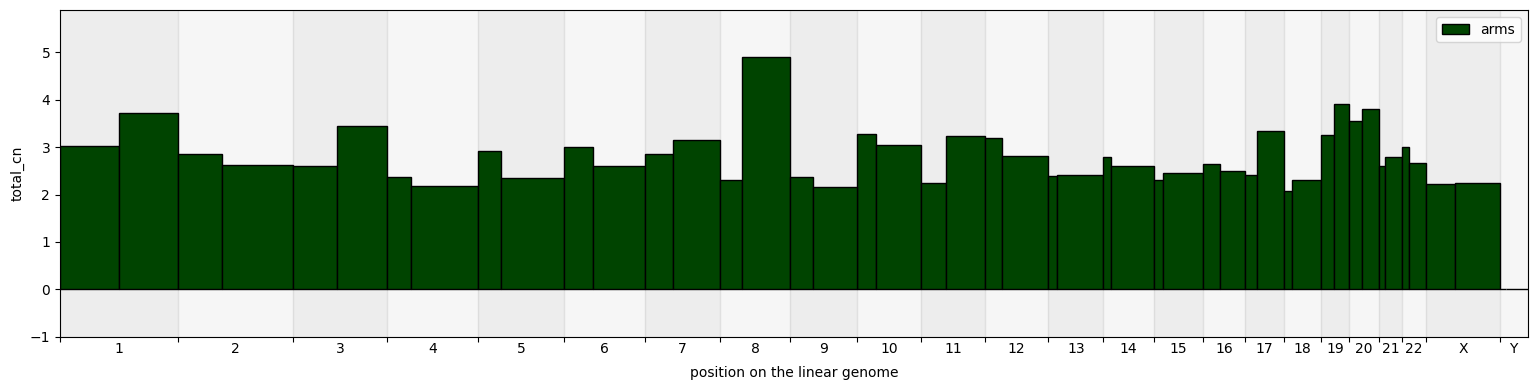
[16]:
cns.fig_bars(pcawg_arms_groups_df, cn_columns="total_cn", colors="#004400");
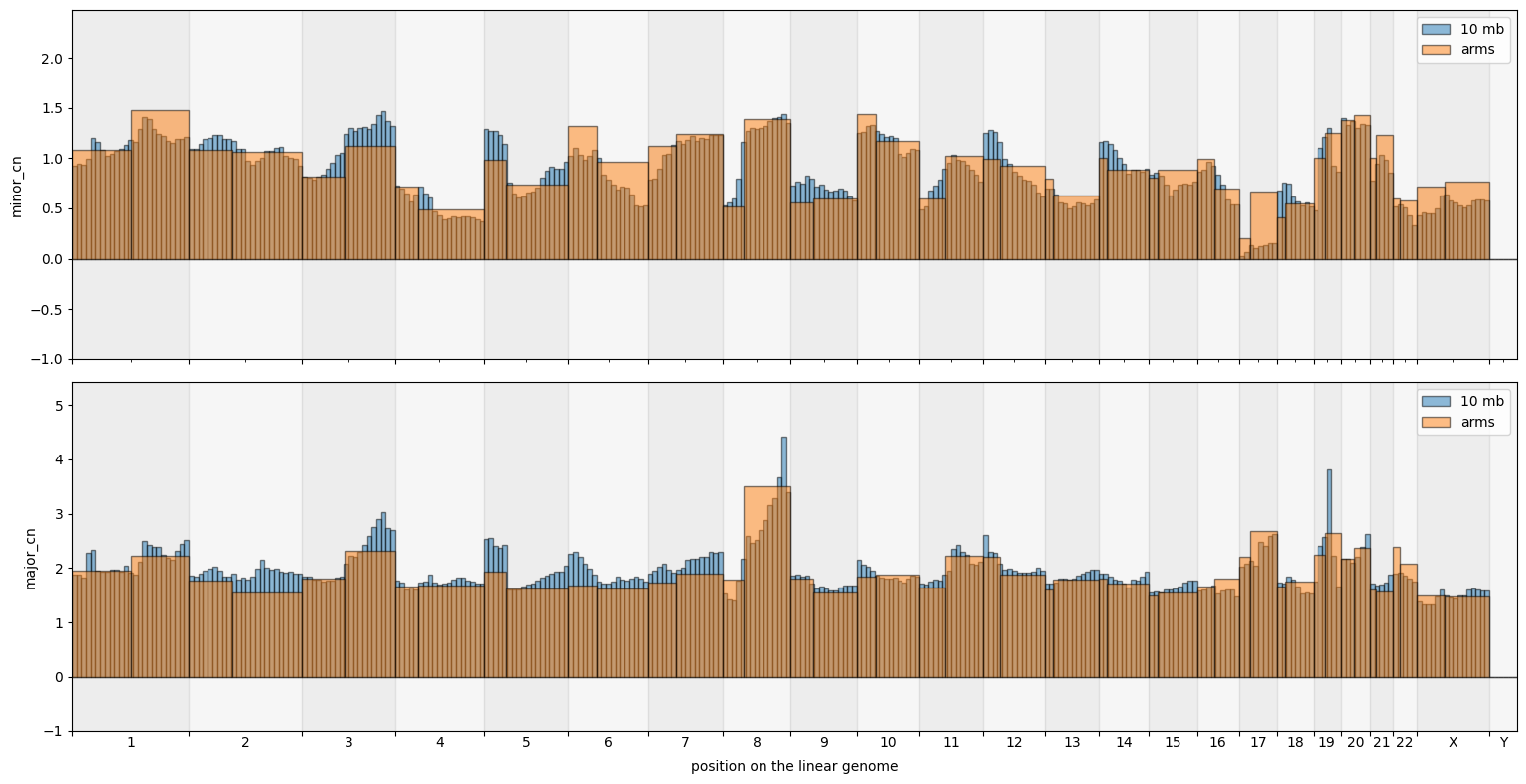
[17]:
big_groups = cns.stack_groups([pcawg_50_groups_df, pcawg_arms_groups_df])
cns.fig_bars(big_groups, cn_columns=["minor_cn", "major_cn"])
[17]:
(<Figure size 1547.84x800 with 2 Axes>,
array([<Axes: ylabel='minor_cn'>,
<Axes: xlabel='position on the linear genome', ylabel='major_cn'>],
dtype=object))
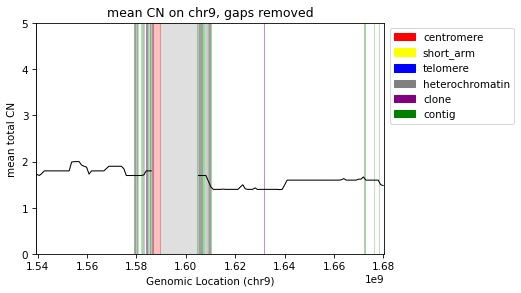
Filter for single chromosome
[18]:
step = int(1e6)
segs = cns.main_segment(remove_segs=cns.make_segments("gaps"), split_size=step, filter_size=step//2)
filtered_cns = cns.main_aggregate(cns.cns_head(pcawg_cns_df, 10), segs=segs, cn_columns="major_cn")
[19]:
grouped_bins = cns.group_samples(filtered_cns).query("chrom == 'chr9'")
fig, ax = plt.subplots(1, 1, figsize=(6, 4), dpi=75)
cns.plot_gaps(ax, alpha=.25, y_max=5)
cns.add_gap_legend(ax)
cns.plot_lines(ax, grouped_bins, "major_cn", color="black")
ax.set_xlim(*cns.x_limits(grouped_bins))
# no_y_ticks(ax)
ax.set_ylabel("mean total CN")
ax.set_xlabel("Genomic Location (chr9)")
ax.set_title("mean CN on chr9, gaps removed")
cdu.save_doc_fig("fig_gaps_removed_chr9")
CN Heatmaps
[20]:
cns.fig_heatmap(pcawg_arms_bin_df, cn_columns="total_cn")
[20]:
(<Figure size 1547.84x300 with 1 Axes>,
<Axes: xlabel='position on the linear genome', ylabel='total_cn'>)
[21]:
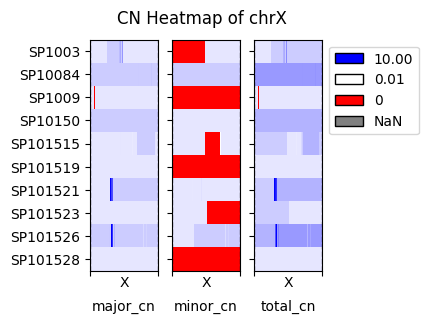
fig, axs = cns.fig_heatmap(pcawg_10_bin_df.query("chrom == 'chrX'"), max_cn=10, vertical=False)
fig.suptitle("CN Heatmap of chrX")
[21]:
Text(0.5, 0.98, 'CN Heatmap of chrX')